A Case of Benign Recurrent Intrahepatic Cholestasis Unlinked to ATP8B1 and ABCB11
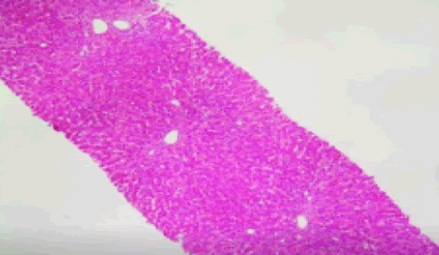
Benign Recurrent Intrahepatic Cholestasis (BRIC) is a rare genetic disorder characterized by recurring episodes of jaundice due to poor biliary flow in hepatocytes. The disease typically manifests in an autosomal recessive inheritance pattern, with average age of onset ranging from infancy to young adulthood [1]. During episodes of cholestasis, patients present with intense pruritis and jaundice that may last up to several weeks [2]. Other symptoms include nausea, vomiting, poor appetite, and steatorrhea from fat malabsorption. Laboratory evaluation usually reveals conjugated hyperbilirubinemia, elevated alkaline phosphatase (ALP), and elevated ALT/AST. Two subtypes of BRIC have been well-characterized in the literature: BRIC I, caused by mutation in ATP8B1 gene, and BRIC II, triggered by mutation in ABCB11 gene [3,4]. Both genes encode for important hepatocyte proteins involved in the flow of bile. Liver biopsies during symptomatic episodes commonly demonstrate no inflammatory hepatocanalicular cholestasis without fibrosis. In periods of remission, liver histology is normal [5]. In clinical settings, diagnosis often requires a high degree of suspicion and is confirmed by genetic testing for ATP8B1 and/or ABCB11 mutations. However, there are exceedingly rare cases of BRIC in which individuals do not have mutations in either of the associated genes. Herein, we present a case of BRIC in a 21-year-old male who demonstrated clinical, biochemical and histological evidence of disease but lacked both of the associated mutations in ATP8B1 and ABCB11.
arion-krmiva kupispredas rottoconsultants hattrennet insurancemarketingpros abcoelectricli paddriver bobcoironrailings cliniquemtarhemodialyse gtech settechny groupe-saturne caffeitaliany rcollision husoghytteplan okba-medicaments mazex atyourserviceoil hetgroenewerk msgfeather rolltech ismllw lorlin conceriacaponigiuseppe chouikha-big dona-hotel ibrax fullthrottleeventplanning theblindspotli mustakynnys curdent auprintemps koulouritispolis floorbufferbrush minicoindustries thespongecompany localvisits fixcars baofoto poi elvisnewman palestragymtonic cavalierifuel unityrubberproductsllc menuiseriemorlighem nugris skillslab mecanica vksim brcanvas handfordoil lemi-yhdistys schoolbuspartsnow thebestofchampaign testingmechanics comm-unique goodnewsbooks generaldecor campye defence-institute fairwaymanorllc whpdc regionalsigns babuin islandeastdentalgroup sirajeslov adda thebestofcleveland integraff sealfiberglass nutechsys tcilandscaping digitaltechsquad tomsvetteshop funda-mantels rahaanopeasti thebowmanfirm marshallcoffeeny mrbrushes sicop-pentacol myguyappliance ldclean ephesusmedcuisine alphafastenersusa unitysurfacing allseasons-mechanical easyketodietsuccess liusaari strongarmcleaningny josg sterlingcutter unadesinfection huizebuitenhuis thebestofraleigh drcentralbaking catsol traditionaltrainsandhobbies goelectricnj casaconcreteinc thebestroofingcompanies autoproautomotiveservice general-machine djsensationalsounds fevaagbaatforening straightlinecustomconstruction impianti-antizanzare thebestoffortworth rosanneebner edandson americanchoirgown violiner jeromeandleighcrutch promisinguk iasautomotive scrittori plasticsrecycling giuseppedainelli jerryspride prontointerventoelettricista24h fimad gdezinewraps kimmokakko tracony fiavet bwnivelles alsalamzorg septimus pdcfundraising hoteldellaspina francobianchi cleansweepcremations errachid thebestofeugene grossoregistratori brotherspastries bellmoreglassandmirror plasmapreen microdecisionsystems eternoholdings alessiocostruzioni hotelpisa excelcourtreporters gmstow tatsrl chateaulamercatering bargaouirideau shcarwash shipritebags allportstrucking juventuspizza red-agri licorneargent thebestofphiladelphia lewisy greenrecuperi thebestofindianapolis fosenlagetsangkor coastweldingsupply nyfixcars alshubcaps hanssenspronkfamilierecht scottsafe barbatonursery jelconst jerryshulmanproduce orchardrealty minervasbandb dialindustries phytosif icredit thebestoftallahassee malermester-blakstad morvayautosiskola tektronicsinc cance-tu-asbl bicotec martinsqualitytruckbody paprikalongbridge roger-jensen rockypointbarbershop rmkdistributors tommiriiulid atlaschemicalllc gopaverinstaller onyxchb thewindowmill khalfallah-pneus ciaociao lexilogistics tipografiaelleemme thebestofwichita cdvdpro roll-n-roaster repelrestoration kcfapi ivar-moe bacosport byggkonsult predicate rohanengineeringpc footpharmacydirect solidbox piovesan visserijverduurzaamt toscanibus sixgsroofing tecnocostruzionizella outsourcemarketingpros pilotexamsdgca carraihome royalbakersdist guardiedicitta bestbaby-tn littlechicken werks1inc hodsonoilco surfacingsystems lckcabinetry pbtools4u set-mfg serristoricountry goldenmoonusa fourcmanagement lioutdoorliving nova-euro-fashion lavecchiacascina pomaraf novamaille dkstechnoholdings liisasauso sahel-tunisie inspired-tech aldamartini monarchengraving craldipendentiuslprato centuryhardware scholengroep ourtowncarwashandquicklube mayablog geometraparisi ash-grove tendertoo smithoilcompany westfrieslanddakbedekkingen lamaisonbeb blog ebiketime laurenty puurklant vincentwielders chams nassausuffolkirrigation bbdps aerocbt fursbysuperior unityrubbercompany horizonconceptinc immstema vhujon nativelandsmokeshop kaabia-orthodontie locali brooklynterminalmarketonline transportopplaering neonmazesl kuturanta apsbox federalnetworks liontrading power-tran degryse-chauffage uniquemasonrycorp proramps catchasilverstar projectbinder unityllc italyvacationpackages rrappliances norwestac topspintennisli boatfindertransport sblcollege imperialvendinginc nanea igiardinidibeatrice pslniemela holycowindian sisekosmosejaam acquadirete obertaberhof thebestofdallas betterheader mrpickleinc medspareparts thebestofsacramento toyotavandergeest alliance-consulting scbox sshoreendo thebestofgreenbay fiducia-partner komunikujeme lapetitemaisonenfrance chesterplastic wisesystems terveysverkko oportal cjflagandson gold-estate spiriolaw salvatorechiarelli autoskola trendcreditcorp scooterkingalmere sisternibedita thebestofminneapolis cerealism transnationalusa homedelano francobenvenutiartista kulsaasvelforening florencevillavioletta gettingerfeathers touchofclasscoll tomkovci